Содержание страницы о миопатии Ландузи-Дежерина
- Общее описание ЛЛПМД (перевод материалов MDA.org)
- Диагностика ЛЛПМД (список клиник и врачей)
- Организации и сообщества, посвящённые ЛЛПМД. Регистры пациентов с ЛЛПМД
- Полезные материалы о ЛЛПМД
- День осведомлённости о ЛЛПМД
- Статьи о ЛЛПМД на нашем сайте
Общее описание мышечной дистрофии Ландузи-Дежерина (Лице-лопаточно-плечевая мышечная дистрофия, ЛЛПМД, FSH, FSHD)
Перевод материалов Muscular Dystrophy Association. Ссылка на источник: https://www.mda.org/disease/facioscapulohumeral-muscular-dystrophy
Что такое лице-лопаточно-плечевая мышечная дистрофия?
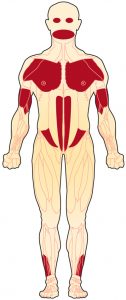 Лице-лопаточно-плечевая мышечная дистрофия (ЛЛПМД, FSH, FSHD, мышечная дистрофия Ландузи-Дежерина, миопатия Ландузи-Дежерина) — это генетическое нервно-мышечное заболевание, при котором мышцы лица, лопаток и верхние части рук наиболее поражены.
Лице-лопаточно-плечевая мышечная дистрофия (ЛЛПМД, FSH, FSHD, мышечная дистрофия Ландузи-Дежерина, миопатия Ландузи-Дежерина) — это генетическое нервно-мышечное заболевание, при котором мышцы лица, лопаток и верхние части рук наиболее поражены.
Английское название заболевания Facioscapulohumeral происходит от трёх латинских слов: facies — медицинский термин для лица; scapula — анатомический термин для лопатки; andhumerus — предплечье и анатомический термин для кости, которая идёт от плеча до локтя. Термин “мышечная дистрофия” означает прогрессирующую дегенерацию мышц с возрастающей мышечной слабостью и атрофией (потеря массы).
При ЛЛПМД лицо, плечи и верхние части рук страдают в первую очередь и наиболее серьёзно, но болезнь, как правило, поражает и другие мышцы.
Симптомы ЛЛПМД
ЛЛПМД обычно начинается в возрасте до 20 лет со слабости и атрофии мышц вокруг глаз и рта, плечевых мышц, мышц верхней части рук и нижней части ног. Позже слабость может распространиться на мышцы живота, а иногда и мышцы бёдер.
Некоторые эксперты делят ЛЛПМД на взрослую и инфантильную формы. Взрослая форма ЛЛПМД (также включает в себя ЛЛПМД, которая начинается в подростковом возрасте) наиболее распространена.
При любом типе ЛЛПМД слабость лицевых мышц может начаться в детстве.
Случается, что и другие симптомы ЛЛПМД появляются в раннем детстве. Мышечная слабость при инфантильной форме ЛЛПМД более ярко выражена, и иногда могут быть поражены слух и зрение. Предполагается, что форма ЛЛПМД с инфантильным началом связана с потерей большой части ДНК.
Для получения дополнительных сведений см. Signs and Symptoms.
Из-за чего появляется ЛЛПМД?
ЛЛПМД может быть унаследована и от отца, и от матери, при этом наследование может произойти, даже если в семейной истории такого заболевания ни у кого не было. Почти всегда заболевание связано с генетическим дефектом (мутацией), из-за которого образуется более короткий сегмент ДНК на хромосоме 4. Сегмент не является частью какого-либо конкретного гена, но, тем не менее, он мешает корректной обработке генетического материала.
В немногих случаях у людей появляется заболевание с симптомами, которые полностью соответствуют ЛЛПМД, но у них нет короткого сегмента на 4-й хромосоме. Генетическая причина такого вида заболевания до сих пор не определена.
Более подробную информацию о генетических причинах ЛЛПМД см. Causes/Inheritance. Более подробную информацию о том, как недостающий сегмент на 4-й хромосоме может привести к ЛЛПМД, см. Research.
Прогрессирование ЛЛПМД
ЛЛПМД обычно прогрессирует очень медленно и редко влияет на сердце или дыхательные пути. Большинство людей с этим заболеванием имеют нормальную продолжительность жизни.
Статус исследований ЛЛПМД
В 2009 году исследователи, поддерживаемые Muscular Dystrophy Association, обнаружили, что фрагменты гена под названием DUX4 аномально активируются в клетках, пострадавших при ЛЛПМД, что приводит к образованию потенциально токсичных белков. Блокирование таких ошибочно активированных генов или белков, образованных в результате работы этих генов, является одним из вероятных путей для лечения ЛЛПМД. Для получения дополнительных сведений см. Research.
Источник: https://www.mda.org/disease/facioscapulohumeral-muscular-dystrophy
Диагностика ЛЛПМД
Общий список клиник и врачей, занимающихся нервно-мышечными заболеваниями, — по ссылке.
Организации и сообщества, посвящённые ЛЛПМД. Регистры пациентов с ЛЛПМД
На сайте TREAT-NMD вы сможете найти подробную информацию о международных организациях, занимающихся лице-лопаточно-плечевой мышечной дистрофией Ландузи-Дежерина: http://www.treat-nmd.eu/FSHD/overview/. Также на этом сайте вы сможете найти международные регистры пациентов.
Почему нужно регистрироваться в реестре?
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку мышечные дистрофии являются редким (орфанным) заболеванием.
Для этого в разных странах ведутся реестры (регистры) пациентов — базы данных по генетической и клинической информации о людях, страдающих нервно-мышечными заболеваниями и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Полезные материалы о ЛЛПМД
Международное сообщество, занимающееся проблемами мышечной дистрофии Ландузи-Дежерина, FSHD Global Research Foundation Ltd. подготовило брошюры с полезной информацией о ЛЛПМД: читать на сайте сообщество (на английском языке).
День осведомлённости о ЛЛПМД
 20 июня — это день осведомлённости о лице-лопаточно-плечевой мышечной дистрофии (мышечная дистрофия / миопатия Ландузи-Дежерина).
20 июня — это день осведомлённости о лице-лопаточно-плечевой мышечной дистрофии (мышечная дистрофия / миопатия Ландузи-Дежерина).
Подробнее о дне осведомлённости о ЛЛПМД:
Статьи о ЛЛПМД на нашем сайте: https://i-mio.org/tags/fshd/