
Данный цикл статей о мышечной дистрофии Беккера собран из открытых источников за 2024 год. Они представляют из себя выдержки из научных статей. К каждому из 3 материалов выпуска приложен оригинал научной статьи для подробного ознакомления. Тема первого выпуска: «Сочетанные заболевания».
Лице-плече-лопаточная мышечная дистрофия 1-го типа в сочетании с мышечной дистрофией Беккера: история болезни одной семьи
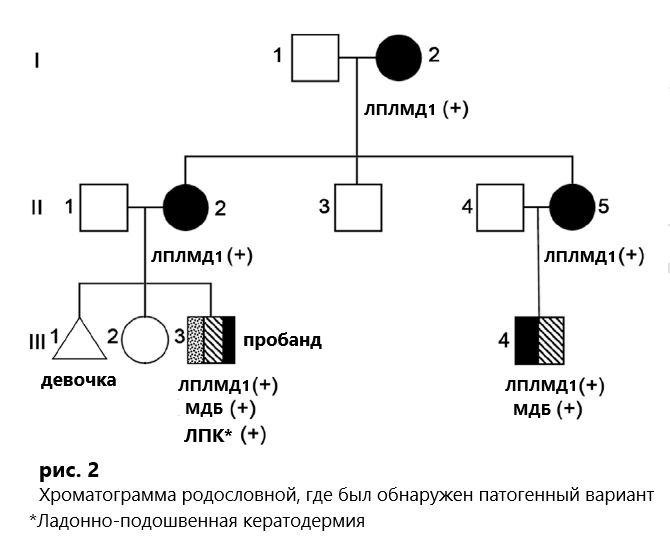
| Лице-плече-лопаточная мышечная дистрофия 1 типа (FSHD1) и мышечная дистрофия Беккера (МДБ) — это разные заболевания, вызванные различными генетическими вариациями и имеющие разный характер наследования. Сочетание обоих заболеваний в одной семье встречается редко. В данном случае речь идет о 10-летнем мальчике, у которого наблюдалась слабость круговой мышцы глаза и рта, плеча и проксимальных отделов верхних и нижних конечностей. Генетическое тестирование показало, что количество повторов D4Z4 в субтерминальной области 4qA хромосомы 4q35 у пациента составляет всего 4 (нормальное значение ≥ 11), и в то же время была обнаружена гетерозиготная делеция в экзонах 13-29 гена DMD у пациента [пробанд*], таким образом, диагноз был клинически и генетически совместим как с FSHD1, так и с МДБ. Исследование родословной показало, что его бабушка по материнской линии, мать, тетя и двоюродный брат также страдали слабость мимических мышц, плеч и конечностей. Генетическое тестирование подтвердило, что у каждого из четырех родственников было по четыре повтора D4Z4 в области 4qA, и все они несли гетерозиготную делецию в экзонах 13-29 DMD. На основании Х-сцепленных признаков МДД/МДБ бабушке по материнской линии, матери и тете был поставлен диагноз FSHD1 в сочетании с носительством делеции в гене DMD, а двоюродному брату по материнской линии — FSHD1 в сочетании с МДБ. В данном исследовании выявлена семья с сочетанным клиническим проявлением FSHD1 и МДБ, что имеет важное справочное значение для диагностики и лечения наследственных миопатий. |
Лечение метилфенидатом китайского мальчика с мышечной дистрофией Беккера в сочетании с синдромом дефицита внимания с гиперактивностью: история болезни
Пациентом был 9-летний мальчик, у которого наблюдались повышенный уровень креатинкиназы в сыворотке крови и снижение концентрации внимания. Магнитно-резонансная томография мышц бедра обеих нижних конечностей показала частичную жировую инфильтрацию ягодичной мышцы с обеих сторон, и была выявлена новая гетерозиготная мутация (c.31 + 6 T > C) в гене МДД методом секвенирования следующего поколения (NGS), а результаты секвенирования были проверены с помощью метода Сэнгера. После тщательного обследования у ребенка также был диагностирован сопутствующий СДВГ, и, учитывая этот новый диагноз, мы начали лечение метилфенидатом в дозе 18 мг/день, и через 6 месяцев лечения у ребенка наблюдалось значительное улучшение концентрации внимания. Мы выявили новую гетерозиготную мутацию в гене МДД, которая расширит спектр патогенных вариантов при МДД. Одновременно лечение метилфенидатом значительно улучшило внимание у детей с МДБ и сопутствующим СДВГ, и это исследование представляет ценность для будущих протоколов лечения. Однако, насколько нам известно, это единственное сообщение о лечении СДВГ с сопутствующим МДБ. Поэтому необходимы дальнейшие исследования для определения взаимосвязи между этими расстройствами и их лечением.
Эпилепсия при мышечной дистрофии Дюшенна и Беккера
В нашем исследовании распространенность эпилепсии была выше, чем в общей популяции (1,4%; 95% доверительный интервал: 0,7-3,2%), но значительно ниже, чем в более мелких выборках пациентов с дистрофинопатией, о которых сообщалось ранее. Не было обнаружено существенных различий в распространенности эпилепсии между МДД и МДБ или в зависимости от генотипов, лежащих в основе заболевания. Когнитивные нарушения не были связаны с более высокой частотой эпилепсии. Наиболее распространенные типы эпилепсии при дистрофинопатиях соответствовали тем, которые наблюдались в более широкой педиатрической популяции, при этом у большинства пациентов эпилепсия эффективно контролировалась с помощью монотерапии. Фактическая распространенность эпилепсии при дистрофинопатиях может быть значительно ниже, чем предполагалось ранее, возможно, вдвое или даже меньше. Наше исследование дает ценное представление об эпилепсии у лиц с дистрофинопатиями, что может повлиять на оказание медицинской помощи.
* пробанд — лицо, с которого начинается составление родословной при генеалогическом анализе.
Выдержки научных статей переведены и собраны врачом-неврологом, председателем правления МОО «Проект Ай-Мио» Бережной Е.Н.
Дополнительные иллюстрации подготовлены: Бережным Д.С.
Изображение к записи составлено из иллюстраций загруженных с freepik.